Perturb-seq Tutorial (R)#
The original data of Jin et al 2020 can be downloaded from the Broad single cell portal (https://singlecell.broadinstitute.org/single_cell/study/SCP1184). Here, we just use a subset of the data to demonstrate the workflow of the analysis.
library(Seurat)
## Loading required package: SeuratObject
## Loading required package: sp
##
## Attaching package: 'SeuratObject'
## The following objects are masked from 'package:base':
##
## intersect, t
sc.seurat <- readRDS("perturbseq-exneu.rds")
# Access the counts through Seurat when its installed version recognizes the
# serialized Assay5 object; otherwise recover the same layer and dimnames
# directly. The fallback keeps this tutorial runnable with older Seurat builds.
if ("RNA" %in% Assays(sc.seurat)) {
counts <- GetAssayData(sc.seurat, assay = "RNA", layer = "counts")
} else {
rna.assay <- sc.seurat@assays[["RNA"]]
counts <- rna.assay@layers[["counts"]]
rownames(counts) <- rownames(rna.assay@features)
colnames(counts) <- rownames(rna.assay@cells)
}
Y <- data.frame(t(as.matrix(counts)), check.names = FALSE) # cell-by-gene matrix
metadata <- sc.seurat@meta.data
perturb <- metadata
colnames(perturb) <- gsub("Perturbation", "trt_", colnames(perturb))
perturb$trt_ <- relevel(as.factor(perturb$trt_), ref = "GFP")
A <- data.frame(
model.matrix(~ trt_ - 1, data = perturb)[, -1, drop = FALSE],
check.names = FALSE
) # cell-by-trt matrix; remove the first (GFP control) column
For running causarray, we require the following inputs:
Y: the cell-by-gene gene expression matrix.A: the cell-by-condition binary matrix of the perturbation/treatment conditions.X, X_A: (optional) the cell-by-covariate matrix of the covariates of interest for outcome and propensity models.
Here, Y and A can be dataframes.
Use R package reticulate to load the Python package causarray.
Render this tutorial from the causarray-r environment defined by
environment-r.yaml; reticulate then connects to the NumPy-2-based
Python causarray environment.
require(reticulate)
## Loading required package: reticulate
Sys.setenv(PYTHONUNBUFFERED = TRUE)
use_condaenv('causarray')
causarray <- import("causarray")
cat(causarray$`__version__`)
## 0.0.8
# (Y, A) should be either data.frame or matrix
# optional covariates can be provided as matrices
dat <- causarray$prep_causarray_data(Y, A)
names(dat) <- c("Y", "A", "X", "X_A")
list2env(dat, .GlobalEnv)
## <environment: R_GlobalEnv>
We first apply gcate to estimate unmeasured confounders.
r <- 10
res_gate <- causarray$fit_gcate(Y, X, A, r, verbose=TRUE) # a list of results from 2 stages optimization
## {'d': 30, 'n': 2926, 'p': 3221, 'r': 10}
## 'Estimating initial latent variables with GLMs...'
## 'Fitting nb GLM (fast)...'
## 'Estimating initial coefficients with GLMs...'
## 'Fitting nb GLM (fast)...'
## {'kwargs_es': {'max_iters': 50,
## 'patience': 5,
## 'rel_tol': 0.0002,
## 'tolerance': 0.0,
## 'warmup': 0},
## 'kwargs_glm': {'disp_glm': array([ 1.11673516, 1.06870944, 1.16716468, ..., 12.58818245,
## 16.46897663, 1.70852614], shape=(3221,)),
## 'family': 'nb',
## 'size_factor': array([0.53193358, 0.87362742, 1.2235467 , ..., 0.5593801 , 0.73025856,
## 0.77857223], shape=(2926,))},
## 'kwargs_ls': {'C': 1000.0,
## 'alpha': 0.1,
## 'beta': 0.5,
## 'max_iters': 20,
## 'recheck_interval': 10,
## 'sparsity_boost': 2.0,
## 'sparsity_threshold': 0.5,
## 'tol': 0.0001,
## 'tol_cell': 0.0001,
## 'tol_gene': 0.0001,
## 'warmup_iters': 0}}
## 'Fitting GCATE (step 1)...'
## {'d': 30, 'n': 2926, 'p': 3221, 'r': 10}
## {'kwargs_es': {'max_iters': 50,
## 'patience': 5,
## 'rel_tol': 0.0002,
## 'tolerance': 0.0,
## 'warmup': 0},
## 'kwargs_glm': {'disp_glm': array([ 1.11673516, 1.06870944, 1.16716468, ..., 12.58818245,
## 16.46897663, 1.70852614], shape=(3221,)),
## 'family': 'nb',
## 'size_factor': array([0.53193358, 0.87362742, 1.2235467 , ..., 0.5593801 , 0.73025856,
## 0.77857223], shape=(2926,))},
## 'kwargs_ls': {'C': 1000.0,
## 'alpha': 0.1,
## 'beta': 0.5,
## 'max_iters': 20,
## 'recheck_interval': 10,
## 'sparsity_boost': 2.0,
## 'sparsity_threshold': 0.5,
## 'tol': 0.0001,
## 'tol_cell': 0.0001,
## 'tol_gene': 0.0001,
## 'warmup_iters': 0}}
## 'Fitting GCATE (step 2)...'
U <- res_gate[[2]]$U
Next, we apply causarray to estimate the causal effects of perturbations on gene expression. Here the 106 GFP control cells and the perturbation groups (median 89 cells) are approximately balanced. We therefore use pooled variance to retain power in this relatively small comparison. This is a dataset-specific choice: unequal variance remains preferable when arm sizes or effective sample sizes are meaningfully unbalanced, when pseudo-outcome variability differs between arms, and for the Replogle and case-control tutorials.
offsets <- log(res_gate[[2]][['kwargs_glm']][['size_factor']]) # use the precomputed size factors
res <- causarray$LFC(Y, cbind(X, U), A, cbind(X_A, U), offset=offsets,
usevar="pooled", verbose=TRUE)
## 'Estimating LFC...'
## {'a': 29, 'd': 11, 'd_A': 12, 'estimands': 'LFC', 'n': 2926, 'p': 3221}
## {'offset': array([-0.63123664, -0.13510128, 0.20175377, ..., -0.58092607,
## -0.31435661, -0.25029351], shape=(2926,)),
## 'random_state': 0,
## 'verbose': True}
## 'Fit propensity score models...'
## {'C': 1.0,
## 'class_weight': 'balanced',
## 'fit_intercept': False,
## 'random_state': 0,
## 'verbose': False}
## 'Fit outcome models...'
## 'Fitting nb GLM (fast)...'
## ('Fast GLM coefficients exceed bound (max|B|=1.63e+05 > 1e+04); falling back '
## 'to statsmodels...')
## 'Estimating dispersion parameter...'
## 'Fitting poisson GLM with offset...'
## 'Fitting nb GLM with offset...'
## 'Fitting GLM done.'
## 'Estimating AIPW mean...'
names(res) <- c("df_res", "estimation")
list2env(res, .GlobalEnv)
## <environment: R_GlobalEnv>
library(dplyr)
##
## Attaching package: 'dplyr'
## The following objects are masked from 'package:stats':
##
## filter, lag
## The following objects are masked from 'package:base':
##
## intersect, setdiff, setequal, union
library(ggplot2)
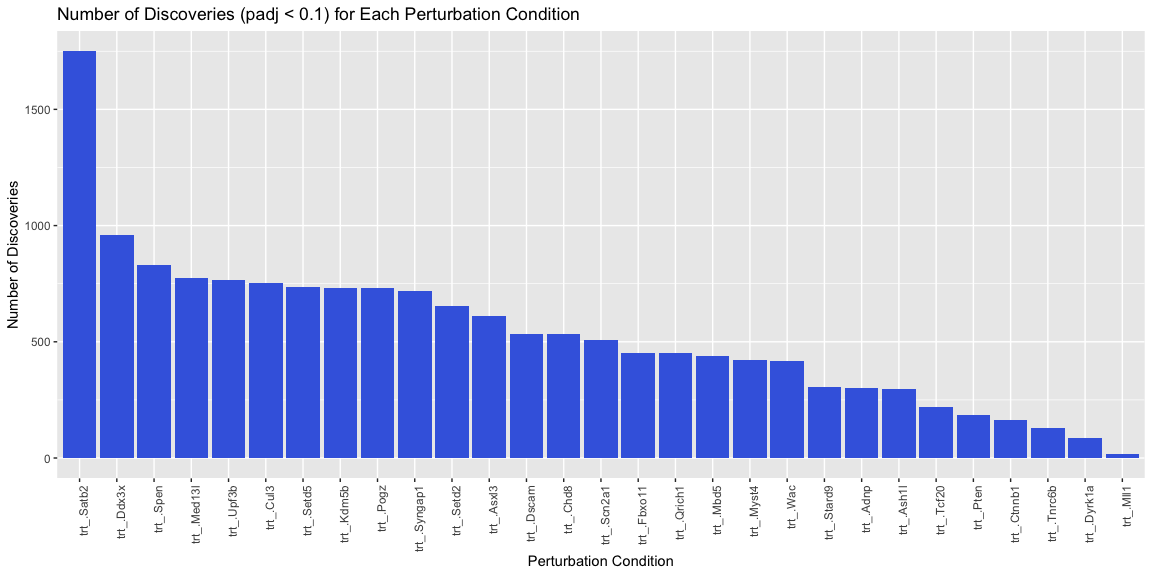
# Filter the results for significant discoveries
significant_discoveries <- df_res[df_res$padj < 0.1, ]
# Count the number of discoveries for each perturbation condition
discovery_counts <- as.data.frame(table(significant_discoveries$trt))
colnames(discovery_counts) <- c('Perturbation', 'Count')
# Order the discovery_counts by Count in descending order
discovery_counts <- discovery_counts %>% arrange(desc(Count))
# Set the factor levels of Perturbation to ensure ggplot respects the order
discovery_counts$Perturbation <- factor(discovery_counts$Perturbation, levels = discovery_counts$Perturbation)
# Plot the number of discoveries for each perturbation condition
ggplot(discovery_counts, aes(x = Perturbation, y = Count)) +
geom_bar(stat = "identity", fill = "royalblue") + theme(axis.text.x = element_text(angle = 90, hjust = 1)) +
ggtitle('Number of Discoveries (padj < 0.1) for Each Perturbation Condition') +
xlab('Perturbation Condition') +
ylab('Number of Discoveries')